
Mutaties in het filamine A gen
De
X-gebonden recessieve vorm werd voor het eerst beschreven door Monteleone en
Fagan in 1969 en door Newbury-Ecob in 1993.
Voor deze studie werd gebruik gemaakt van een klinisch en genetisch
gekarakteriseerde grote familie (familie 1a) die is aangedaan met X-gebonden
myxomateuze klepdystrofie (XMVD) (OMIM 314400).
Met behulp van linkage analyse werd het gen gemapt tot een 8-cM interval op chromosoom Xq28. Een groot familiaal en genealogisch nazicht leidde tot de verfijning van de locus op het XMVD gen en standaard positie klonen identificeerde een P637Q mutatie in het filamine A (FLNA) gen bij alle aangedane familieleden. Screening van extra, kleinere, niet verwante families met ernstige vormen van klepdystrofie lieten toe nog drie andere mutaties in dit gen te vinden. De huidige studie toont aan dat filamine A het eerste bekende gen is verantwoordelijk voor geïsoleerde niet syndromale klepaandoeningen.
Methode
Klinische evaluatie
De studie werd uitgevoerd volgens de Franse richtlijnen voor genetisch onderzoek
met toestemming van het ethisch comité van het universitair ziekenhuis van
Nantes.
De klinische evaluatie omvatte een anamnese en een klinisch onderzoek met nadruk
op het cardiovasculaire systeem en enige bindweefselziekten. De fenotypering van
familieleden was gebaseerd op echocardiografie en de gebruikte data zijn deze
van het meest recente onderzoek, met uitzondering van de patiënten die
klepchirurgie ondergingen. Transthoracale echocardiogrammen werden gemaakt
volgens de criteria van de American Society of Echocardiography. Metingen van de
mitralisklep werden gedaan op de parasternale, lange as met 2D beelden. De
lengte van elk klepblad werd bepaald onmiddellijk voor sluiting van de klep. De
dikte van het vrije uiteinde van de mitralisklepbladen werd gemeten binnen een
geselecteerd diastolisch frame dat de mitralisklepbladen en chordae duidelijk
uiteen hield. Men ging ervan uit dat mitralisklepprolaps aanwezig was, wanneer
tweedimensionale opnames in het parasternale lange as aanzicht een protrusie
toonde van de mitralisklepbladen in het linker atrium voorbij de lijn tussen de
annulaire scharnierpunten en wanneer het coaptatiepunt van de klepbladen ter
hoogte van of boven het annulaire vlak blijft tijdens systole.
Mitralisklepregurgitatie werd geschat met de standaardmethodes waaronder de PISA
(proximale isovelociteit oppervlakte gebied) analyse. Aortaklepregurgitatie werd
beschouwd als aanwezig te zijn, wanneer een abnormale diastolische flow vanuit
de aortaklep in de linker ventriculaire outflow tractus werd gevonden. Beelden
van de tricuspidalisklep werden gemaakt in vierkameraanzicht en de
pulmonalisklep werd geanalyseerd in het hoge linker parasternale korte as zicht.
Patiënten werden gedefinieerd als aangedaan, wanneer de dikte van het vrije
uiteinde van een of beide mitraliskleppen meer was dan 4 mm, met of zonder
mitralisklepprolaps en mitralisklepregurgitatie op het echocardiogram vanwege de
inconsistentie bij aangedane individuen. Omdat aortaklepdystrofie moeilijk te
bepalen is op transthoracale echocardiografie werd de regurgitatie
gekwantificeerd. Patiënten werden tevens beschouwd als aangedaan in geval van
milde tot ernstige aortaklepregurgitatie.
Genetische
analyse
Voor moleculaire studies werd DNA geïsoleerd uit perifeer bloed (lymfocyten) en
uit paraffinegebonden klepweefsel volgens de standaard methode.
Linkage
analyse, fijnere mapping en haplotype constructie
Familie 1 was reeds gelinkt aan chromosoom Xq28. Om de kandidaatregio te
verkleinen werden 8 microsatellietmarkers (DXS998, DXS8091, DXS8069, DXS8061,
DXS15, DXS1073, F8 en DXS1108) van de kandidaat geselecteerd om te gebruiken bij
linkage analyse in de uitgebreide familie. F8 is een dinucleotidemarker binnen
factor VIII genintron 13. Er werd een tweepuntslinkage analyse gedaan met het
FASTLINK programma. Voor de linkage berekeningen werd aangenomen dat er sprake
was van een X-gebonden overerving met een ziekte allelfrequentie van 0,0001 en
2% fenokopieën. De penetrantie werd gezet op 100% voor de mannelijke
familieleden en op 70% voor de vrouwelijke familieleden. Alle familieleden
werden meegenomen in de analyse. Patiënten werden gedefinieerd als aangedaan
wanneer de dikte van de mitralisklep meer was dan 4 mm en wanneer milde tot
ernstige aortaklepregurgitatie aanwezig was. Personen met een mitralisklepdikte
minder dan 2 mm zonder aortaklepregurgitatie werden gedefinieerd als niet
aangedaan en personen met een klepdikte tussen de 2 en 4 mm werden beschouwd als
‘fenotype onbepaald’. Om meer recombinaties te vinden en de locus te verfijnen
werden 11 microsatellietmarkers (DXS8103, DXS1684, BXS10052, DXS10053, DXS10054,
Afm308yhl, DXS10051, DXS10049, Afm0l82xa5, GABRA3 en DXS10047) gegenotypeerd bij
de recombinante individuen om de centromerische grens te bepalen. Twee intragene
microsatellietmarkers, GAB3 en F8, werden gebruikt om de telomerische grens te
bepalen.
Analyse van
kandidaatgenen en mutatieanalyse van FLNA
Kandidaatgenen in het Xq28 interval werden gescreend op mutaties met behulp van
sequentie analyse bij een aangedaan persoon van de familie 1a. Coderende exonen
en short flanking intronsequenties werden geamplificeerd met PCR en overmatige
primer werd van de geamplificeerde fragmenten verwijderd met exoSAP en
gesequenteerd met een dye-terminator cycle-sequencing systeem. De coderende
sequentie van FLNA (exonen 2 tot 48) werden geamplificeerd van genoom DNA met
behulp van primers zoals hiervoor beschreven.
Varianten van FLNA geïdentificeerd met de sequentie analyse werden bevestigd met
restrictie-enzymdigestie met HaeIII en Sau96I. De benodigde exonen werden
geamplificeerd met PCR, gedigesteerd met restrictie-enzymen en in stukjes
geknipt op een 8% acrylamide gel. In de twee gevallen bleek de variant een
restrictieplaats minder te hebben. De V711D mutatie werd bevestigd door
afgeleide gekliefde geamplificeerde polymorfe sequencing. Om de frequentie van
imitaties na te gaan werd gebruik gemaakt van sequentieanalyse,
restrictie-enzymdigestie of afgeleide gekliefde geamplificeerde polymorfe
sequencing met DNA van familieleden en van 500 controle chromosomen van
Europese, Afrikaanse, of Aziatische origine.
Modelbouw
Om de structurele consequenties van de geïdentificeerde mutaties te bestuderen
werd een driedimensionaal model geconstrueerd van humaan filamine. Dit
geautomatiseerde homologe model werd gemaakt met het programma INSIGHTII.
Resultaten
Klinische evaluatie
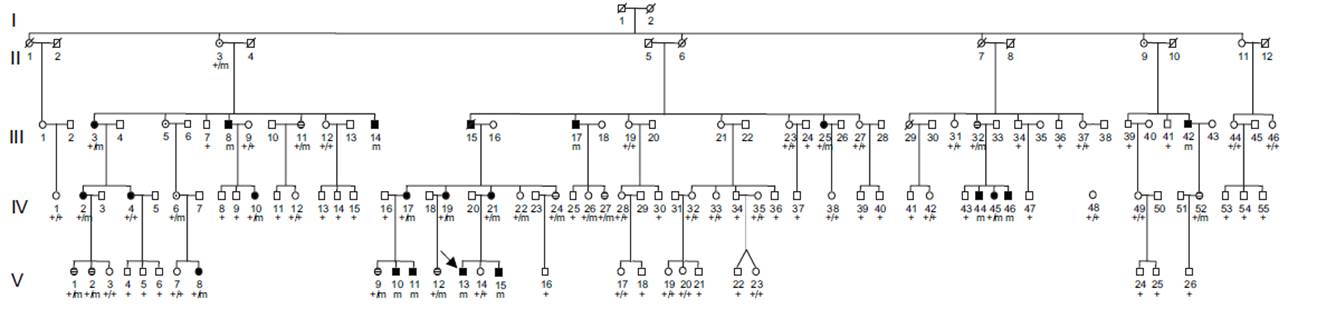
Familie 1a is en eerder vermelde blanke Franse familie die is aangedaan met XMVD
en bestaat momenteel uit 91 levende leden. De testpersoon (patiënt V-13)
onderging een aortaklepvervanging vanwege rustige regurgitatie op de leeftijd
van 17 jaar. De afmetingen en morfologie waren normaal en klinisch onderzoek
bracht geen bindweefsel- of gewrichtsabnormaliteiten aan het licht.
Hartauscultatie gaf een aortisch regurgitatie geruis en echocardiografie toonde
ernstige regurgitatie. Op MRI bleken de dimensies van de thoracale aorta normaal
en niets duidde op aspecten van Marfan of Ehlers Danlos. Histologisch onderzoek
van de klep toonde typische kenmerken van myxomateuze klepziekte met duidelijke
verdikking van het vrije uiteinde van de klep.
De neef van de testpersoon (patiënt V-10) onderging een valvuloplastie voor
ernstige regurgitatie te wijten aan mitralisklep dystrofie op de leeftijd van 20
jaar. Detectie van een milde hemofilie A bij deze twee patiënten en een
familiestudie leidden tot de identificatie van een zeer grote familie van meer
dan 300 individuen. Onder de 44 mannelijke familieleden hebben er 10
progressieve mitralisklepprolaps, die in vier gevallen is geassocieerd met
middelmatige tot ernstige aortische regurgitatie en waarvan er 4 klepchirurgie
ondergingen. Van de 47 vrouwelijke familieleden konden allen worden beschouwd
als aangedaan met mitralisklep-of aortaklepabnormaliteiten, ofschoon allen
asymptomatisch waren. Een kind (patiënt V -8), gediagnosticeerd op de leeftijd
van 10 jaar, toonde ernstige aortische regurgitatie met aortaklep stenose. Bij
alle aangedane familieleden was de klepziekte geassocieerd met een milde
hemofilie A (Factor VIII activiteit tussen 15% en 50%).
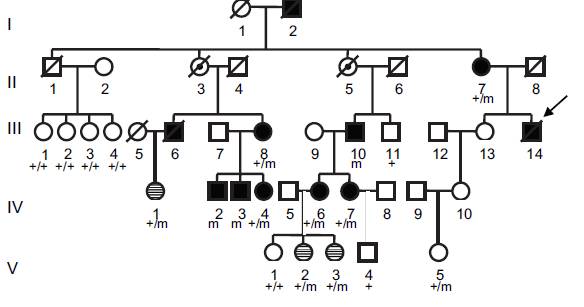
 Figuur 3. Familie 2
|
Familie 2 was een Britse familie met XMVD. De testpersoon (patiënt IV-1)
was geboren met ernstige aangeboren klepziekte en overleed 24 uur na de
geboorte aan ernstig hartfalen. Autopsie toonde dystrofie van alle vier
de kleppen en een atriumseptumdefect. Zijn grootvader (patiënt II-2)
onderging een mitralisklep vervanging en sluiting van een persisterend
foramen ovale op de leeftijd van 41 jaar. Tijdens de chirurgie bleken de
kleppen myxomateus en dystrofisch te zijn. Hij was gediagnosticeerd met
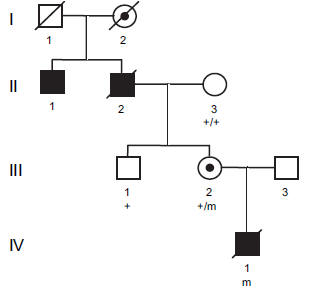
mitralisklep- en aortaklepziekte op de leeftijd van 30 jaar. Het eerste kind van gezonde zwarte Afrikaanse ouders (familie 3) was een jongetje. Antenataal was hij gediagnosticeerd met abnormaal dikke hartkleppen op foetale echocardiografie en werd geboren op 38 weken in goede gezondheid. Postnatale echocardiografie bevestigde middelmatige tricuspied incompetentie, triviale mitralisklep en pulmonalisklep incompetentie, en milde aortaklep incompetentie. Alle kleppen waren verdikt en dystrofisch. Op de leeftijd van vier maanden was zijn groei en ontwikkeling normaal en toonde hij geen kenmerken van cardiaal falen. Een echocardiografie toonde een uitstekende ventriculaire functie. De mitralisklep bleef dystrofisch zonder bewijs van regurgitatie en er was een zeer milde aortaklepregurgitatie. Zijn moeder was klinisch onderzocht en toonde geen spoor van hartafwijking.
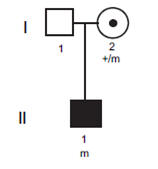
Familie 4 was van Chinese origine. De twee jongens, 4 en 12 jaar oud,
hadden beide mitralisklep en aortaklep dystrofie. Bij de patiënt II-1
werd en hartgeruis gevonden op de leeftijd van vier maanden tijdens een
routineonderzoek. Aanvullende echocardiografie toonde aan dat hij een
polyklepziekte had met myxomateuze verdikking van de mitralisklep,
tricuspidalisklep en aortaklep. Hij had een significante mitralisklep-
en tricuspidalisklepregurgitatie en milde aortaklepregurgitatie. Patiënt
II-2 werd geïdentificeerd door het verhaal van zijn broer. Ook hij bleek
de polyklepziekte te hebben. |
|
 Figuur 4. Familie 3
|
 Figuur 5. Familie 4
|
|
Linkage analyse en mapping ter inperking van de locus
Een significante linkage (Zmax=8,6 bij θ=0 voor DXS1108) werd verkregen voor
familie 1. Om de XMVD locus te verfijnen werden additionele markers
gegenotypeerd. Recombinatie op DXS10049 voor de niet aangedane mannelijke
patiënt IV-54 van familie 1a levert de centromerische grens van de locus.
Analyse van markers van leden van familie 1b toont hetzelfde ziekte haplotype,
met uitzondering van patiënt IV-3. Deze persoon die is aangedaan met
klepdystrofie, maar met een normale coagulatiefactor activiteit had een
recombinatie van
microsatellietmarker
GAB3, hetgeen de telomerische grens van de gelinkte regio aanduidt en aantoont
dat de valvuloplastie en hemofilie onafhankelijk werden overgedragen. Het
ziektegen bevindt zich dus ergens tussen DXS10049 en GAB3, een interval van 2,5
Mb, dat niet het factor VIII gen bevat.
Mutatie identificatie
Na uitsluiting van kandidaatgenen in het ziekte interval en verscheidene
voorspelde genen, werd FLNA onderzocht door middel van directe sequencing. Het
is opgebouwd uit 48 exonen, ongeveer 26 Kb, distaal grenzend aan het emerinegen.
Mutatie analyse van alle coderende regionen van FLNA bij de aangedane leden van
familie 1 toonde aan dat allen een C-aan-A transitie hebben van nucleotide 1910
in exon 13. Dit voorspelt een missense mutatie substitutie van proline (P) voor
glutamine (Q) in aminozuur 637. Bij familie 1 zijn in totaal 13 aangedane
mannelijke familieleden en 30 vrouwelijke familieleden mutatiedragers. Een
klinisch aangedane vrouw (patiënt IV-4) erfde de mutatie niet en bleek een
fenokopie te zijn. Een G-aan-A transitie van nucleotide 862 in exon 5, wat duidt
op een missense mutatie van glycine (G) voor arginine (R), werd gevonden bij de
aangedane leden van familie 2. Bij het aangedane lid van familie 3 was er een
T-aan-A transitie van nucleotide 2132 in exon 14 wat leidt tot valine (V) voor
aspartaamzuur (D) in aminozuur 711. De relaties in sequentie werden niet
gevonden bij niet aangedane familieleden of bij de 500 controlechromosomen van
zowel blanken als zwarte Afrikanen.
De mutaties werden bevestigd door te testen op verlies van een HaeIII (P637Q) en
Sau96I (G288R) restrictieplaats respectievelijk de gederiveerde gekliefde en
geamplificeerde polymorfe sequentie (V711D. Tot slot werd een 1944-bp
genoomdeletie gevonden coderend voor exonen 16 tot 19, overeenkomend met de
546-bp coderende sequentie (NT_025965.13:g.942144_944086del1944insTG) die een
in-frame deletie van 182 residuen voorspelt (van V761 tot Q943) bij twee jongens
(familie 4) gediagnosticeerd met polyklepdystrofie. De deletie werd
gevisualiseerd met PCR en sequencing reactie van intron 15 tot 19 om aan te
tonen dat aangedane individuen een kleiner PCR product hebben, hetgeen
overeenkomt met een deletie van 1944 bp. Deze deletie werd niet gevonden in 200
controlechromosomen, waaronder 100 Aziatische chromosomen.
Naast de mutaties geïdentificeerd in FLNA werden ook andere gekende
polymorfismen gevonden bij de patiënten. Deze één-nucleotide polymorfismen
traden op met relatief normale frequenties en sommigen ervan resulteerden in
aminozuursubstitutie. Er werden geen andere ziekte geassocieerde varianten
gevonden. Screening van drie andere families met potentieel X-gebonden
valvulaire dystrofie toonde geen mutaties in FLNA.
Genotype-fenotyperelaties in familie 1
Relaties bij mannelijke
familieleden
In familie 1
waren 13 mannen drager van de P637Q mutatie. Alle mannelijke dragers hadden
mitralisklep problemen en twee van hen hadden reeds chirurgie ondergaan hiervoor
op de leeftijd van 20 respectievelijk 51 jaar. Op een na hadden alle mannelijke
dragers mitralisklepprolaps. Vier van hen hadden een prolaps van het anterieure
klepblad, acht van hen van beide klepbladen. Geen van hen had een geïsoleerde
prolaps van het posterieure klepblad. De diktes van de mitraliskleppen bedroegen
5,3±1,1 mm voor het anterieure klepblad en 4,4 ±0,9 mm voor het posterieure
klepblad. Op een na hadden allen tevens aortaklepregurgitatie. In zes gevallen
mild was deze, bij drie matig en bij drie ernstig, dus leidend tot
aortaklepvervanging. Er waren geen afwijkingen aan de aortaboog en alle
aortakleppen waren tricuspied. De leeftijden van aortaklepvervanging lagen
tussen de 17 en 52 jaar. Bij 11 mannelijke dragers werd milde tot middelmatige
tricuspidalisregurgitatie gevonden en milde pulmonalisregurgitatie bij vier
dragers. Voor deze problemen was echter geen chirurgie noodzakelijk.
Relaties bij vrouwelijke
familieleden
Geen van de 30
heterozygote vrouwen was symptomatisch en geen onderging klepchirurgie. Onder de
vrouwelijke dragers van de P637Q mutatie werden er 14 als aangedaan beschouwd,
12 twijfelgevallen vanwege mineure klepziekte en 4 als niet aangedaan. In vier
gevallen werd mitralisklepprolaps gevonden, waarvan bij drie gevallen het
anterieure klepblad was aangedaan en bij een het posterieure. Onder de
heterozygote vrouwen hadden er 10 een milde mitralisklepregurgitatie en 4 een
middelmatige. De klepdiktes bedroegen 3,3±0,5 mm voor het anterieure klepblad en
2,8±0,7 mm voor het posterieure klepblad. De klepbladen waren dus bij de mannen
significant dikker dan bij de vrouwen. Acht heterozygote vrouwen hadden een
milde aortaklepregurgitatie, drie een middelmatige en een een ernstige. Negen
heterozygote vrouwen hadden een milde tricuspidalisklepregurgitatie, een een
middelmatige. Twee heterozygote vrouwen hadden een milde
pulmonalisklepregurgitatie, een een middelmatige.
Structurele aspecten van
de FLAN mutaties bij XMVD
Sequentievergelijkingen toonden aan dat alle drie de missense mutaties
modificaties zijn van sterk geconserveerde residuen waarvan er minstens tien ver
teruggaan in de evolutie en dientengevolge bij vele gewervelden voorkomen.De
G288R, P637Q, en V711D mutaties zijn gelokaliseerd binnen de eerste, de vierde
en de vijfde repeat en leiden tot een kleiner eiwit dat repeats 5 tot 7 mist.
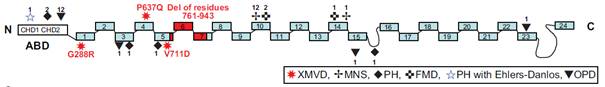
Figuur 6. Schematische weergave van het bindend domein van een filaminemolecuul met aanduiding van gestoorde punten
![]()
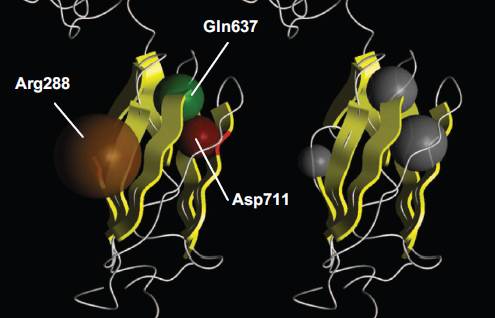
Figuur 7. Model van humaan filamine. De antiparallelle β-ketens zijn geel en de gekleurde bollen stellen uitvergroot de al dan niet foutieve aminozuren voor.
Het voorspelde driedimensionale model van humaan filamine bestaat uit zeven antiparallelle strengen gelegen in twee vlakken. In het model is te zien dat het Gly288 residu gelegen is op het externe oppervlak, terwijl de Pro637 en Val711 residuen intern gelegen zijn. Voor alle mutanten geldt dat de vervanging van een apolair residu voor een polair residu (G288R basisch, P637Q ongeladen en V711D zuur) leidt tot een verhoogde polariteit en dus een significante verandering in de structurele confirmatie van de strengen teweeg brengt. Alle drie de substituties samen leiden tot verstoring van de proteïne-ligand-receptor binding.
Bespreking
In deze studie werd de identificatie van FLNA als het eerste verantwoordelijke
gen voor een niet syndromale klepdystrofie besproken. De identificatie van dit
genetische defect, overgedragen volgens een X-gebonden recessief patroon, werd
ondersteund door een aanpak met genealogische en geografische oogpunten. Uit het
onderzoek is bewijs voortgekomen dat aantoont dat de P637Q mutatie in het
FLNA
gen verantwoordelijk is
voor het klepdefect in familie 1. Linkage analyse leverde positieve LOD-scores
op en alle aangedane mannen zijn dragers van de mutatie. Daarnaast leverde
screening van extra ongerelateerde families met klepdystrofie nog drie andere
mutaties van het FLNA gen (G288R, V711D, en een 182 aminozuurdeletie). Geen van
deze mutaties werd gevonden in de 500 controle chromosomen. Alle missense
mutaties bevonden zich in de regio coderend voor filamine. Binnen de families
met XMVD werd de ziekte overgeërfd met complete penetrantie bij mannen en
incomplete penetrantie bij vrouwen met variabele gradaties in expressie
consistent met verschillende X-chromosoom inactivatie patronen. Onder de 13
mannelijke dragers van de P637Q mutatie, hadden er 12 een typische
mitralisklepprolaps gekarakteriseerd door verdikte klepbladen en geassocieerd
met milde tot ernstige aortaklepregurgitatie en in 11 gevallen
tricuspidalisklepregurgitatie. Onder de 30 vrouwen waren er 14 aangedaan (zoals
te verwachten viel ongeveer de helft). De vrouwelijke dragers hadden wel dikkere
kleppen dan de normale vrouwen, maar waren toch minder ernstig aangedaan dan de
mannen (geen van hen onderging klepchirurgie).
De leeftijd voor klepchirurgie varieerde tussen de 17 en 52 jaar in familie 1.
De diagnoseleeftijd was sterk variërend en lag binnen familie 1 tussen de 11 en
55 jaar. Bij familie 2 waren twee patiënten gediagnosticeerd met polyklepziekte
op de leeftijden 25 en 30 jaar en eentje bij geboorte. Bij familie 3 werd
polyklepziekte reeds antenataal gediagnosticeerd en op 4 en 12-jarige leeftijd
bij familie 4.
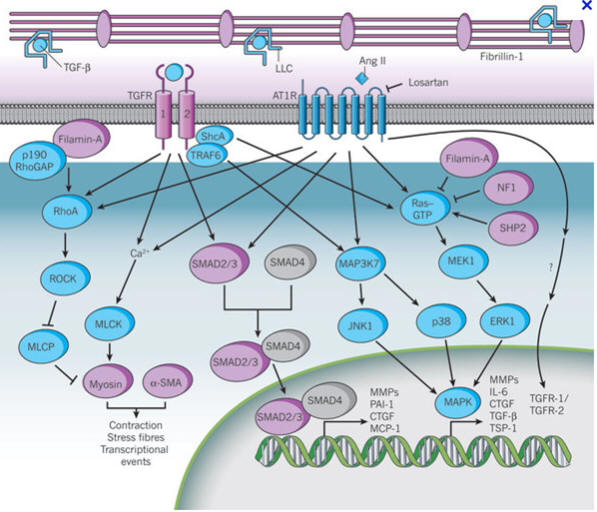
Figuur 8 De TGF-β signaaltransductiepathway.
Filamine
A
is een fosfoproteïne dat
actinefilamenten kruiselings verbindt en het actine cytoskelet aan het
plasmamembraan bindt door interactie met actine en membraanproteïnes zoals
β-integrines. Filamine bestaat uit een actinebindend domein aan de N-terminus en
24 homologe repeats die de ruggengraat van het proteïne vormen. Iedere repeat
bestaat uit 7 antiparalelle β-ketens die in 2 β-vlakken gerangschikt zijn.
Filamines bestaan in vivo als dimeren gemedieerd door interacties tussen de
C-terminale sequenties.
Naast zijn rol als structuurcomponent van het cytoskelet heeft filamine A ook
een rol in de regulatie van diverse signaaltransductiepathways in en rond de
cellen. Het kan bijdragen aan de ontwikkeling van myxomateuze klep veranderingen
door de regulering van TGF-β en aldus leiden tot volledig ontaarde reactie
cascades zoals teruggevonden in ontwikkelingsmodellen. Bij muizen blijkt dat
filamine interageert met de TGF-β en BMP signaalinhibitor Smad6. Gebrek aan
Smad6 bleek te leiden tot hyperplasie van de hartkleppen. BMP6-BMP7
dubbelmutanten blijken echter weer hypoplastische kleppen te hebben.
De verzwakte kleppen kunnen vervormd raken door hemodynamische stress, hetgeen
bepalend is voor de leeftijd waarop de klachten tot uiting komen. De hogere
drukken die optreden in de linker caviteiten kunnen verklaren waarom zowel de
aortaklep als de mitralisklep dikwijls zijn aangedaan. Pulsflow heeft een
welbekend effect op celmorfologie en samenstelling van de extracellulaire
matrix. Bindweefselcellen in mechanisch actief milieu overleven toegepaste
fysische krachten door modificatie van een actine cytoskelet dat de celmembraan
beter stabiliseert. Een andere verklaring is dat modificatiegenen de
progressiegraad van de ziekte kunnen beïnvloeden.
Vervorming van mitraliskleppen kan veroorzaakt worden door defecten in proteïnes
van de extracellulaire matrix gelijkaardig aan de cytoskelet eiwitten en
signaaltransductiepathways die transmissie mediëren tussen de extracellulaire
matrixproteïnes en het cytoskelet bij syndromale klepdystrofie. Identificatie
van nieuwe genen die mitralisklepprolaps veroorzaken laten toe deze hypothese te
bevestigen.
Afzonderlijke mutaties van de filaminegenen produceren verschillende fenotypes.
Mutaties in de
FLNB and
FLNC
genen werden gevonden bij
skeletale en musculaire stoornissen. Mutaties in het FLNA gen werden eerder
toegeschreven aan humane periventriculaire nodulaire heterotopie,
otopalatodigitale syndromen, frontometafyseale dysplasie en Melnick-Needles
syndroom. Klepziekte werd vermeld bij frontometafyseale dysplasie en
periventriculaire nodulaire heterotopie met Ehlers-Danlos. Al deze voorgenoemde
aandoeningen werden in de families van deze studie niet gevonden. Omdat er geen
extracardiale aandoeningen werden gevonden, kan gesteld worden dat het in dit
onderzoek gaat om een geïsoleerde vorm en niet om een onderdeel van een
syndroom.
De mechanismes waardoor verschillende mutaties van FLNA leiden tot een variabele
expressie van de ziekte blijven vooralsnog onbekend. Een groot aantal mutaties
bij periventriculaire heterotopie zijn nonsense en splicing mutaties en alle
mutaties die zijn geïdentificeerd bij de andere aandoeningen zijn missense
mutaties die zich clusteren in kleine regio’s van het proteïne zonder een
duidelijk genotype-fenotype correlatie. De verschillende fenotypes kunnen duiden
op verschillende interacties van filamine met een bepaalde bindingspartner.
![]()